Фенилкетонурията е вродено разстройство. При повечето нелекувани пациенти заболяването се проявява с известна степен на умствена изостаналост. Какво е фенилкетонурия? Прочетете за това в статията..
Съдържание
 Фенилаланинът се отнася до незаменими аминокиселини (вещества, които се използват за изграждане на протеини в човешкото тяло), тъй като животинските тъкани не са в състояние да го синтезират. В човешкото тяло, където фенилаланинът постъпва с храната, той се окислява с помощта на специфичен ензим фенилаланин хидроксилаза до напълно несъществена аминокиселина - тирозин. Нарушаването (блокирането) на тази реакция, наблюдавано в нарушение на синтеза на фенилаланин хидроксилаза в черния дроб, води до развитие на тежко наследствено заболяване - фенилкетонурия.
Фенилаланинът се отнася до незаменими аминокиселини (вещества, които се използват за изграждане на протеини в човешкото тяло), тъй като животинските тъкани не са в състояние да го синтезират. В човешкото тяло, където фенилаланинът постъпва с храната, той се окислява с помощта на специфичен ензим фенилаланин хидроксилаза до напълно несъществена аминокиселина - тирозин. Нарушаването (блокирането) на тази реакция, наблюдавано в нарушение на синтеза на фенилаланин хидроксилаза в черния дроб, води до развитие на тежко наследствено заболяване - фенилкетонурия.
Тирозинът се използва от организма за синтезиране на хормони на щитовидната жлеза. А меланинът, производно на тирозин и фенилаланин, осигурява пигментация (цветно оцветяване) на кожата, очите и косата.
Фенилкетонурията се нарича още фенилаланинемия, фенилпирувична олигофрения. Заболяването се отнася до вродени метаболитни нарушения (метаболизъм), характеризира се с повишаване нивото на фенилаланин в кръвната плазма и е придружено от умствена изостаналост. Точната причина за умствена изостаналост при PKU е неизвестна, но се дължи на биохимичен дефект. Наследствената природа на заболяването се среща при повечето хора. Честотата на PKU е 1 случай на 6000-10 000 новородени.
Симптоми на фенилкетонурия
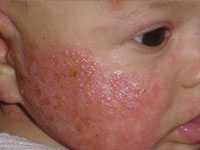
Клиничните симптоми на фенилкетонурия при новородени обикновено липсват, поради което трябва да се извърши лабораторен скрининг (диагностика). В редки случаи се забелязва летаргия и затруднения в храненето. При повечето нелекувани пациенти заболяването се проявява в известна степен на умствена изостаналост (по-често изразена), което е основният симптом на фенилкетонурията. Пациентите обикновено имат по-светла кожа, коса и очи от здравите си роднини. Някои деца могат да имат кожни промени, които приличат на детска екзема.
Пациентите с фенилкетонурия се характеризират с изобилие от неврологични симптоми, рефлексите са особено променени. При по-големи деца се отбелязват както големи, така и малки епилептични припадъци, аномалии в електроенцефалограмата се отбелязват в 75 - 90% от случаите. Развива се подчертано повишаване на активността и психоза, често неприятно, излъчвано от пациентите "мишка" миризма, причинена от наличието на фенилоцетна киселина в урината и потта.
Диагностика на фенилкетонурия
След като новороденото е хранено с мляко (източник на фенилаланин) в продължение на поне 48 часа, лекарите правят скринингов тест за фенилкетонурия (изследване на капка кръв). На възраст 4-6 седмици продуктът на разпадане на фенилаланин може да бъде открит в урината на болно дете. По това време се извършва друг скринингов тест за фенилкетонурия. Изследване на урина се извършва след края на неонаталния период и деца от семейства, в които има пациент с фенилкетонурия, трябва да се повтарят редовно през първата година от живота.
Лечение с фенилкетонурия
Лечението се състои в ограничаване на диетичния прием на фенилаланин, за да се задоволи нуждата от тази незаменима аминокиселина, но не повече; осигуряват нормален растеж и развитие, предотвратявайки натрупването на фенилаланин и крайни продукти от неговия метаболизъм в организма. Необходим е постоянен мониторинг на нивото на фенилаланин в кръвта на детето. В Съединените щати лофеналакът се използва широко за заместване на млякото, което е пълноценно във всички отношения и съдържа малко количество фенилаланин. Позволено е да се ядат естествени продукти с ниско съдържание на протеини, като плодове, зеленчуци, някои зърнени храни и т.н. Нуждата от фенилаланин се задоволява чрез консумация на ограничено количество естествен протеин и остатъчно количество фенилаланин в лофеналак. Вече се предлагат продукти, които са напълно пречистени от фенилаланин. Продуктът с фенилни картофи съдържа всички необходими съставки с изключение на фенилаланин, не съдържа мазнини, поради което енергийната му стойност е по-ниска от тази на други продукти.
Лечението от лекар трябва да се извършва от първите дни от живота. Само ранното и последователно лечение предотвратява увреждане на централната нервна система и тежка умствена изостаналост. Ако лечението започне след 2-3 години от живота, с негова помощ е възможно само да се ограничи изразената повишена активност и да се улесни протичането на конвулсивния синдром..