Кой е изложен на амиотрофична странична склероза? Как протича това заболяване? Авторът на тази статия знае отговорите на тези въпроси..
Съдържание
Амиотрофична странична склероза: разпространение и рискови фактори
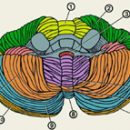
 Амиотрофичната латерална склероза (известна още като болест на моторните неврони, болест на Шарко, в англоговорящите страни - болест на Лу Гериг) е бавно прогресиращо, нелечимо дегенеративно заболяване на нервната система с неизвестна досега етиология.
Амиотрофичната латерална склероза (известна още като болест на моторните неврони, болест на Шарко, в англоговорящите страни - болест на Лу Гериг) е бавно прогресиращо, нелечимо дегенеративно заболяване на нервната система с неизвестна досега етиология.
Болестта се характеризира с прогресивно увреждане на двигателните неврони, придружено от парализа (пареза) на крайниците и мускулна атрофия. В края на пътуването пациентите умират от отказ на дихателните мускули. Амиотрофичната странична склероза трябва да се разграничава от така наречения синдром на ALS, който може да придружава заболявания като енцефалит, пренасян от кърлежи..
Амиотрофичната странична склероза е описана за първи път през 1869 г. от Жан-Мартин Шарко.
Понастоящем причините за развитието на амиотрофична латерална склероза не са точно определени..
По-голямата част от случаите не са свързани с наследственост и не могат да бъдат обяснени положително от никакви външни фактори (минали заболявания, наранявания, екология и др.).
Всяка година 1-2 от 100 000 души развиват амиотрофична странична склероза. Обикновено болестта засяга хора на възраст между 40 и 60 години. От 5 до 10% от пациентите са носители на наследствената форма на амиотрофична латерална склероза; на тихоокеанския остров Гуам е идентифицирана специална, ендемична форма на болестта.
Как протича амиотрофната странична склероза
Ранните симптоми на заболяването: потрепване, спазми, изтръпване на мускулите, слабост в крайниците, затруднено говорене също са характерни за много по-често срещани заболявания, поради което диагнозата на амиотрофична латерална склероза е трудна - докато болестта се развие до степента на мускулна атрофия.
В зависимост от това кои части на тялото са засегнати на първо място, те разграничават:
- Амиотрофичната странична склероза на крайниците (до три четвърти от пациентите) започва по правило с поражение на единия или двата крака. Пациентите се чувстват неловко при ходене, скованост на глезена, спъване. По-рядко се наблюдават лезии на горните крайници и е трудно да се извършват нормални дейности, които изискват гъвкавост на пръстите или силата на ръката.
- Булбарна амиотрофична странична склероза се проявява с трудности при говорене (казва пациентът «в носа», назален, лошо контролира силата на говора, по-късно има затруднения при преглъщане).
Във всички случаи мускулната слабост постепенно обхваща все повече и повече части на тялото (пациентите с булбарна форма на амиотрофична странична склероза може да не доживеят до пълна пареза на крайниците).
Симптоми на амиотрофична странична склероза включват признаци на увреждане както на долния, така и на горния двигателен нерв:
- увреждане на горните двигателни нерви: хипертоничност на мускулите, хиперрефлексия, така наречения анормален рефлекс на Бабински;
- увреждане на долните двигателни нерви: мускулна слабост и атрофия, конвулсии, неволни фасцикулации (потрепвания) на мускулите.
Рано или късно пациентът губи способността да се движи самостоятелно. Болестта не засяга умствения капацитет, но води до тежка депресия в очакване на бавна смърт. В по-късните стадии на заболяването са засегнати дихателните мускули, пациентите изпитват прекъсвания в дишането, рано или късно животът им може да бъде подкрепен само чрез изкуствена вентилация на белите дробове и изкуствено хранене. Обикновено от откриването на първите признаци на амиотрофична латерална склероза до смъртта отнема от шест месеца до няколко години. Известният астрофизик Стивън Хокинг (роден 1942 г.) обаче е единственият известен пациент с еднозначно диагностицирана амиотрофична латерална склероза (през 60-те години), чието състояние се стабилизира с течение на времето..